Immune-mediated diseases, their relationships and divisions
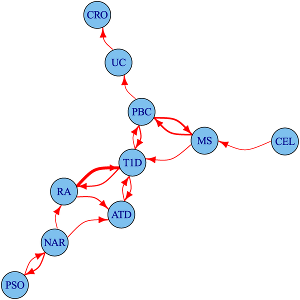
We study the shared and distinct genetic aetiology between related autoimmune diseases both at the genomewide and region specific level, addressing two separate questions. First, at a the level of the whole genome, do associations with one disease predict associations with another disease? We do this using an approach which estimates conditional false discovery rates for association of each disease at each SNP conditional on the other disease. Because this uses genomewide associations, it includes information from individual loci that are not genomewide significant, but show a trend to shared association for two diseases. However, because genes with similar function are often located close to one another, and because genetic variants also show a spatial correlation due to linkage disequilibrium, this does not autoimattically imply that the two diseases share the same causal variants.

To address this more specific question, we therefore use a complementary approach, applying the an adapted colocalisation method that we use for matching disease and gene expression signatures to investigate sharing of causal variants in detail. Our analysis of four autoimmune diseases identified 90 regions that were associated with at least one disease, 33 (37%) of which were associated with 2 or more disorders. For 14 of these 33 overlapping regions, we found evidence that the causal variants differed. This shows that even for two apparently closely related autoimmune diseases which both show association to the same genetic region (delineated by recombination hotspots), this will relate to them sharing the same causal variant only about 50% of the time.

More recently, we have developed a genome-wide test for determining whether some phenotypically or clinically defined division of patients corresponds to a genetic signature of aetiological heterogeneity. This is particularly useful when direct comparison of the two subtypes yields no individual genomewide significant signal. We investigate subgroups of type 1 diabetes (T1D) cases defined by autoantibody positivity, establishing evidence for differential genetic architecture with thyroid peroxidase antibody positivity, driven generally by variants in known T1D associated regions.
Related papers
- Fortune M. Nat Genet 2015
- Liley J. Nat Genet 2017
- Burren O. Genome Medicine 2020
- Ortiz-Fernández L. American Journal of Human Genetics 2020
- Onengut-Gumuscu S. Nat Genet 2015
- Liley J. PLoS Genet 2015
Causal variants
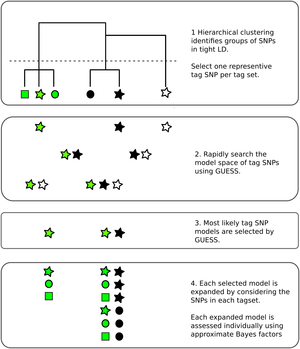
Genetic association studies have identified many DNA sequence variants that associate with disease risk. By exploiting the known correlation that exists between neighbouring variants in the genome, inference can be extended beyond those individual variants tested to identify sets within which a causal variant is likely to reside. However, this correlation, particularly in the presence of multiple disease causing variants in relative proximity, makes disentangling the specific causal variants difficult. Statistical approaches to this fine mapping problem have traditionally taken a stepwise search approach, beginning with the most associated variant in a region, then iteratively attempting to find additional associated variants. We adapted a stochastic search approach that avoids this stepwise process and is explicitly designed for dealing with highly correlated predictors to the fine mapping problem. We showed in simulated data that it outperforms its stepwise counterpart and other variable selection strategies such as the lasso.
Fine mapping results are often used to generate credible sets of SNPs, which are supposed to contain the true causal variant with a specified probability. We have shown that frequentist coverage of these sets is often higher than expected, because this procedure is generally performed conditional on a small p value being seen. Anna Hutchinson has presented this work in various forums, with slides here.
In more recent work, we exploited the established relationships between immune mediated diseases, borrowing information from any other available diseases to increase the resolution of fine mapping results. We highlighted in particular the potential for joint tagging where a single variant, in LD with two causal variants, can be mis-identified by stepwise methods, and showed how stochastic search, and multi-disease fine mapping, can avoid this.
We validated these results using allele specific expression studies of IL2RA in CD4+ T cells. All results are made available in an interactive form for six immune-mediated diseases: autoimmune thyroid disease, celiac disease, childhood arthritis, MS, RA, T1D.
Related papers
- Hutchinson A. Human Molecular Genetics 2020
- Wallace C. PLoS Genet. 2015
- Asimit J. Nat. Commun. 2019
- Hernández N. Nature Communications 2021
Causal genes
Genomewide association studies (GWAS) have been hugely successful in identifying associations between genetic variation and risk of common diseases. However, genetic variation will not be a pharmaceutical target for these disease and translating this knowledge into understanding the genes, cells and pathways involved in disease aetiology has been slow. This is because the associated genetic variants do not typically reside in genes and change the protein they encode, but lie between genes and are presumed to regulate their expression in some cells, perhaps under specific conditions.
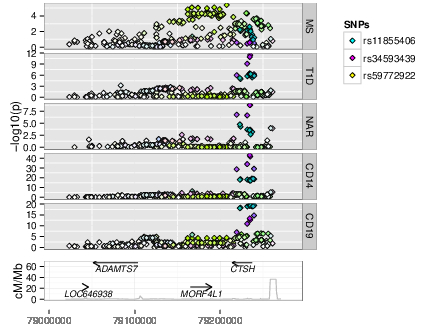
As GWAS studies for disease have been conducted over the past decade, so people have conducted GWAS for other, more gene specific traits, such as parallel GWAS for the expression levels of each of the ~20,000 protein coding genes in a given cell type, called “eQTL” studies. These produce a local GWAS trace just as a disease GWAS does. We use a pattern matching technique called colocalisation to determine whether the two GWAS traces are compatible with the two traits, disease risk and expression of a given gene in a given cell type and condition, sharing a causal variant.
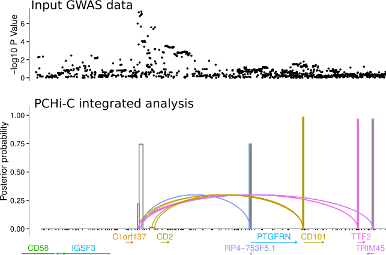
Another approach is to exploit knowledge about the 3D folding of chromatin derived from high throughput Chromosome Conformation Capture (Hi-C) in its targetted form: Capture Hi-C (CHi-C). This allows us to link GWAS causal variants (mapped probabilistically) to the genes they regulate, and we have deployed this approach across 17 primary human sorted cell types , as well as a separate more detailed comparison using CD4+ T cells, both activated and non-activated (1).
Related papers
- Burren O. Genome Biology 2017
- Eijsbouts C. bioRxiv 2018
- Javierre B. Cell 2016
- Burren O. Bioinformatics 2014
- Giambartolomei C. PLoS Genet 2014
- Guo H. Hum Mol Genet 2015
- Martin P. Nat. Commun. 2015
- Wallace C. Genet Epidemiol 2013
Gene expression in immune cells
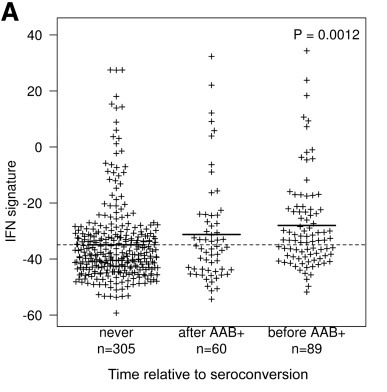
The BABYDIET study led by Annette Ziegler and Ezio Bonifacio collected blood samples longitudinally from 109 children genetically at risk of, but initially unaffected by type 1 diabetes. We were lucky to have access to white blood cells (PBMCs) from these samples and measured the gene expression in them. We were particularly interested in expression of interferon responsive genes, given previous links between type 1 diabetes and infection. Indeed, we saw an upregulation of these genes in children who went on to develop the autoantibodies that are strongly predictive of T1D diagnosis, but, crucially, the upregulation was transient and preceded the appearance of autoantibodies. It was also temporally correlated with recent upper respiratory infection, and may represent a biomarker for the response to infection or the mechanism by which the infection influences type 1 diabetes risk.
We also used these data to investigate seasonal variation in gene expression, and found that 25% of the genes expressed in these cells in these children varied in their expression throughout the year. In winter, we saw that this expression profile produced a pro-inflammatory environment. This might be advantageous during a season when infectious diseases are at a peak, but is a risk factor for other diseases associated with inflammation such as cardiovascular also peak in winter. We replicated this finding in multiple datasets, including one from the Southern hemisphere when winter occurs during June-August.
Related papers