Coloc: using variant-specific priros in coloc
Jeffrey Pullin
2025-12-04
Source:vignettes/a07_variant-specific-priors.Rmd
a07_variant-specific-priors.RmdVariant-specific priors in coloc
## This is coloc version 6.0.1
data(coloc_test_data)
data(eqtlgen_density_data)
attach(coloc_test_data)
attach(eqtlgen_density_data)By default coloc assumes that all variants in a region are equally likely to be causal for the traits of interest. However, there is substantial evidence that functional annotations and other genetic information can provide evidence about whether variants are causal.
Using general variant-specific information
To provide variant-specific information to coloc use the
prior_weights1 or prior_weights2 arguments for
coloc.abf(), coloc.susie() or
coloc.bf(). The arguments specify non-negative weights that
encode the ‘weight’ (proportional to the probability of a variant being
causal). Specifying weights for either or both of the traits will
automatically change the prior probability of a variant being causal for
both traits.
For example, if we had prior information that the first 100 variants were twice as likely to be causal for trait 1 we could encode this in the following prior weights:
## PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
## 8.26e-19 2.91e-10 5.16e-12 8.18e-04 9.99e-01
## [1] "PP abf for shared variant: 99.9%"## Coloc analysis of trait 1, trait 2##
## SNP Priors## p1 p2 p12
## 1e-04 1e-04 1e-05##
## Hypothesis Priors## H0 H1 H2 H3 H4
## 0.892505 0.05 0.05 0.002495 0.005##
## Posterior## nsnps H0 H1 H2 H3 H4
## 5.000000e+02 8.262373e-19 2.911768e-10 5.156170e-12 8.179198e-04 9.991821e-01An example of variant-specific information
Pullin and Wallace (2024) describe the implementation of
variant-specific priors in coloc and compare various sources of prior
information. The best performing source of prior information was the
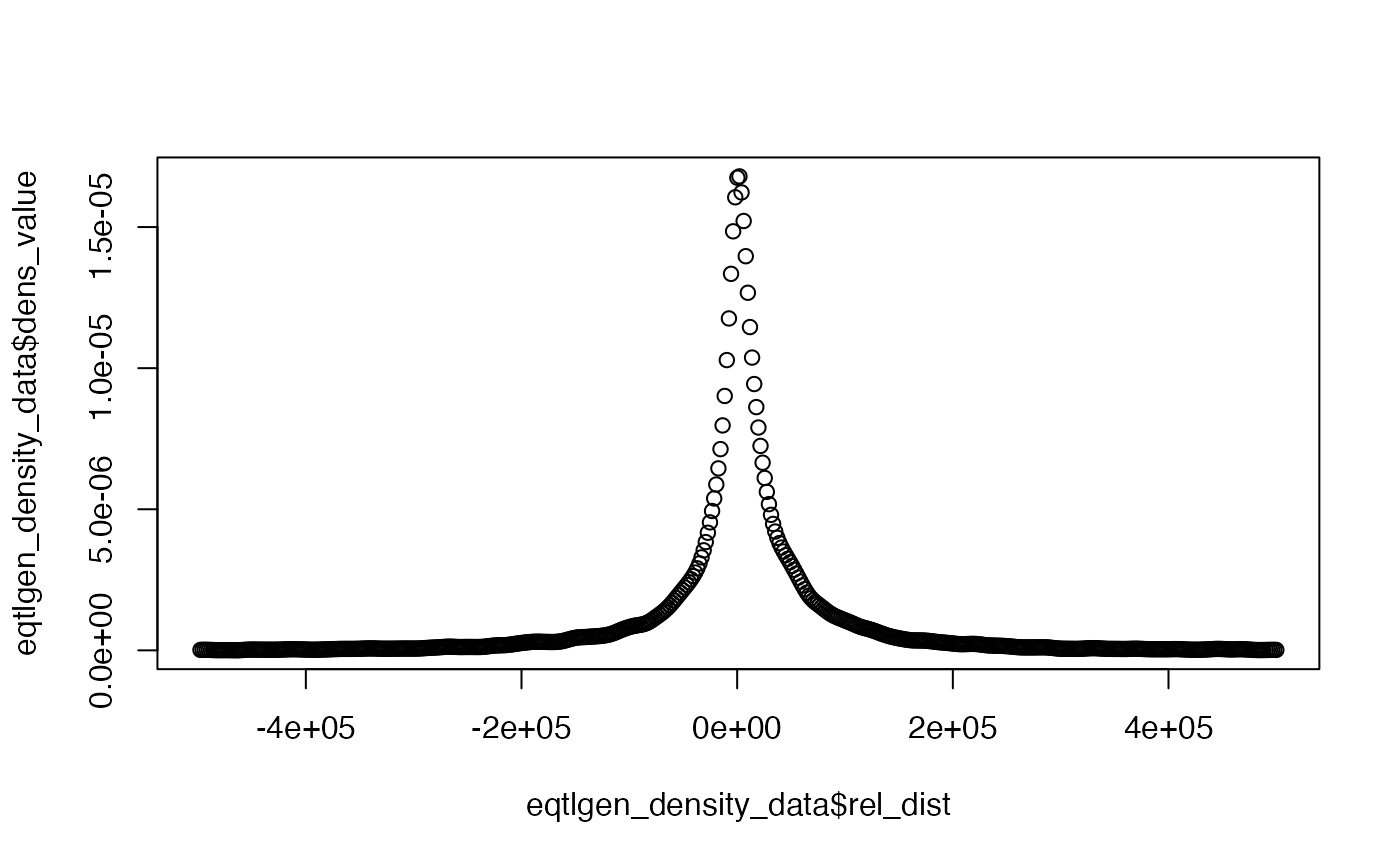
density of the distance between an expression quantative trait loci
(eQTL) and the gene’s transcription start site (TSS). These estimated
densities show that eQTLs often lie close to the TSS, suggesting that we
should expect variants that lie closer to the TSS to be more likely to
be causal. The density estimated from the eQTLGen dataset, see Pullin
and Wallace (2024) for details, is stored in the coloc dataset
eqtlgen_density_data and we can visualise the density,
where the middle of the interval is the TSS.
plot(
eqtlgen_density_data$rel_dist,
eqtlgen_density_data$dens_value
)
To illustrate the use of the TSS we augment coloc’s simulated data with simulated genomic positions in a 1Mb window around the TSS of PTPN22. Then using a simple function we can compute prior weights for these positions relative to different specified TSSs.
pos <- sample(113371759:114371759, 500, replace = FALSE)
D1$position <- pos
D2$position <- pos
compute_eqtl_tss_dist_weights <- function(pos, tss, density_data) {
rel <- pos - tss
closest <- numeric(length(pos))
for (i in seq_along(pos)) {
closest[[i]] <- which.min(abs(density_data$rel_dist - rel[[i]]))
}
out <- density_data$dens_value[closest]
out <- out / sum(out)
out
}Computing the weights and applying them shows little impact on the probability of colocalisation because this simulated example has very strong evidence for colocalisation. In real data, the impact will likely be greater.
# Compute the weights relative to the PTPN22 TSS.
w1 <- compute_eqtl_tss_dist_weights(D1$position, 113871759, eqtlgen_density_data)
coloc.abf(D1, D2)## PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
## 1.38e-18 2.94e-10 8.59e-12 8.34e-04 9.99e-01
## [1] "PP abf for shared variant: 99.9%"## Coloc analysis of trait 1, trait 2##
## SNP Priors## p1 p2 p12
## 1e-04 1e-04 1e-05##
## Hypothesis Priors## H0 H1 H2 H3 H4
## 0.892505 0.05 0.05 0.002495 0.005##
## Posterior## nsnps H0 H1 H2 H3 H4
## 5.000000e+02 1.377000e-18 2.937336e-10 8.593226e-12 8.338917e-04 9.991661e-01
coloc.abf(D1, D2, prior_weights1 = w1)## PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
## 4.61e-17 1.19e-09 2.88e-10 6.44e-03 9.94e-01
## [1] "PP abf for shared variant: 99.4%"## Coloc analysis of trait 1, trait 2
##
## SNP Priors## p1 p2 p12
## 1e-04 1e-04 1e-05##
## Hypothesis Priors## H0 H1 H2 H3 H4
## 0.892505 0.05 0.05 0.002495 0.005##
## Posterior## nsnps H0 H1 H2 H3 H4
## 5.000000e+02 4.607613e-17 1.191318e-09 2.875401e-10 6.440915e-03 9.935591e-01